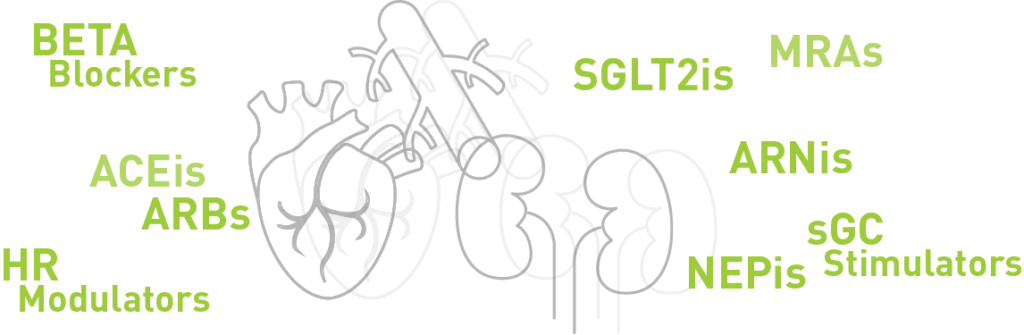
The role of calcitropes in treating heart failure with reduced ejection fraction (HFrEF)
Calcitropes improve contractility but at the risk of side effects, such as worsening energetics or malignant arrhythmias.2
Calcitropes are a class of traditional inotropes that are generally reserved for advanced heart failure as treatment for cardiogenic shock, bridge to LVAD or transplant, or as palliative therapy because long-term use is associated with increased mortality in patients with heart HFrEF.2
The promise of myotropes and mitotropes
In contrast to calcitropes, myotropes and mitotropes may improve long-term myocardial contractility and mortality without increasing cardiomyocyte calcium fluxes or adverse events such as worsening cardiac energetics, raising diastolic pressure, or heart rate.2
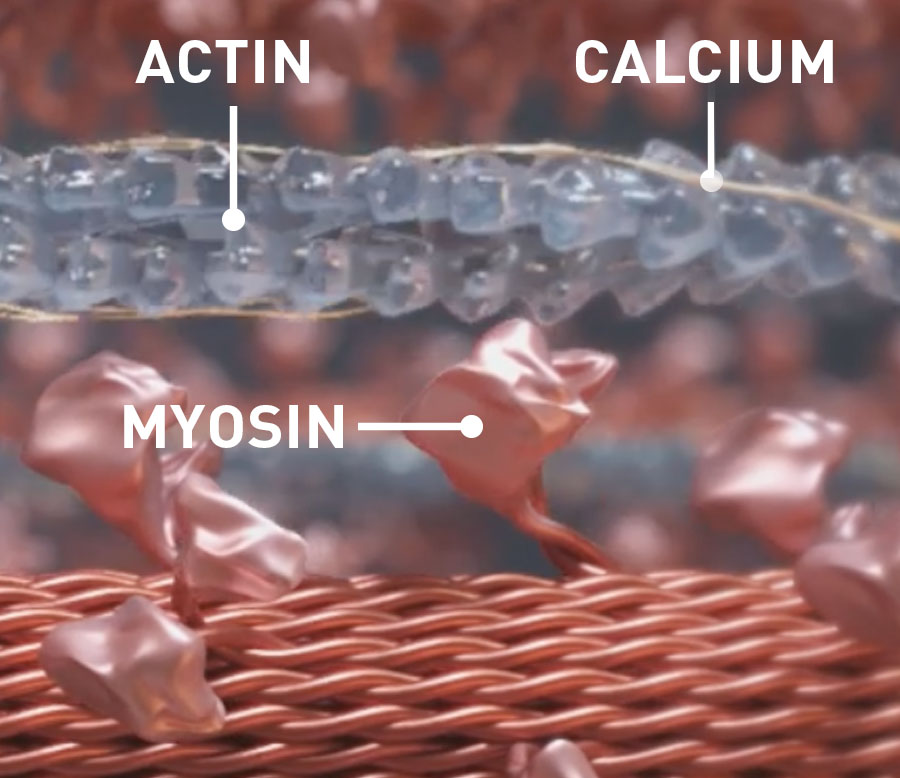
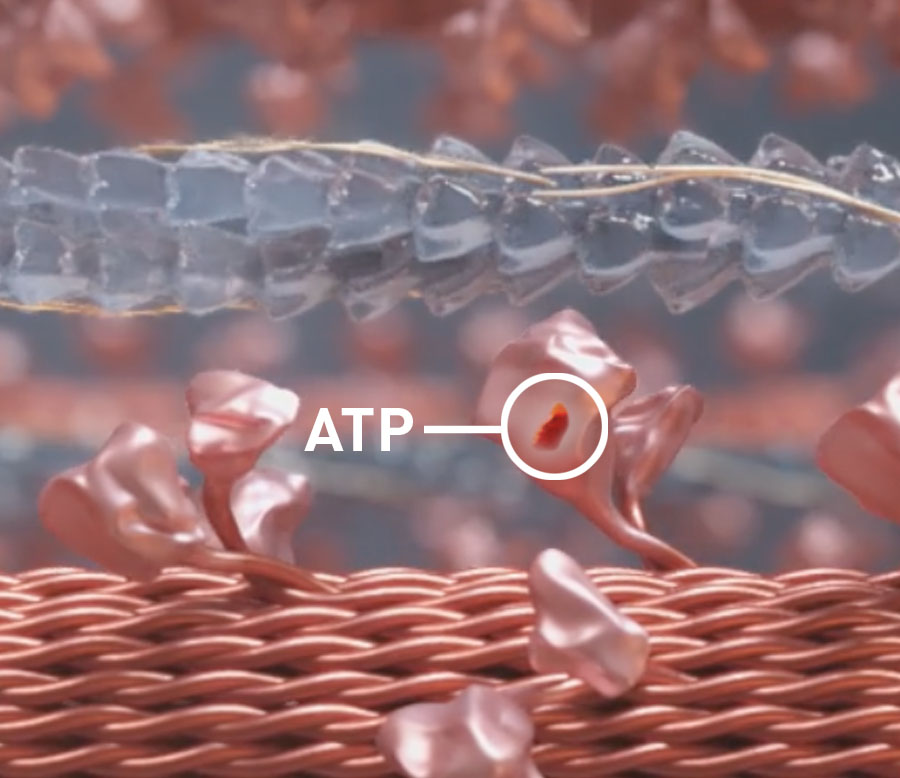
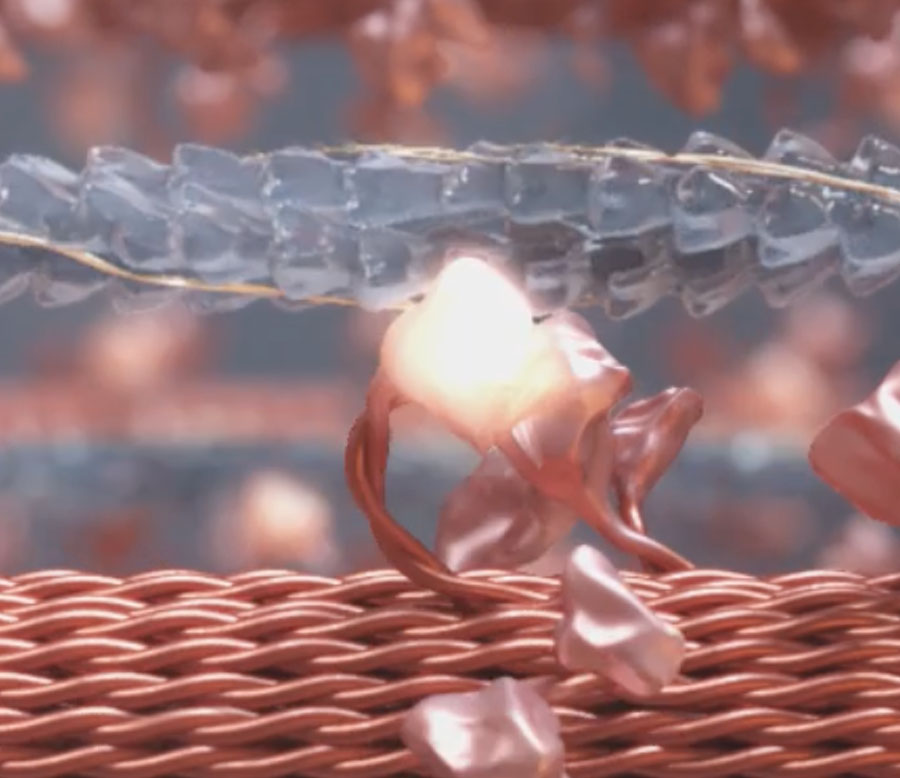
Myotropes’ effect on cardiac energy1
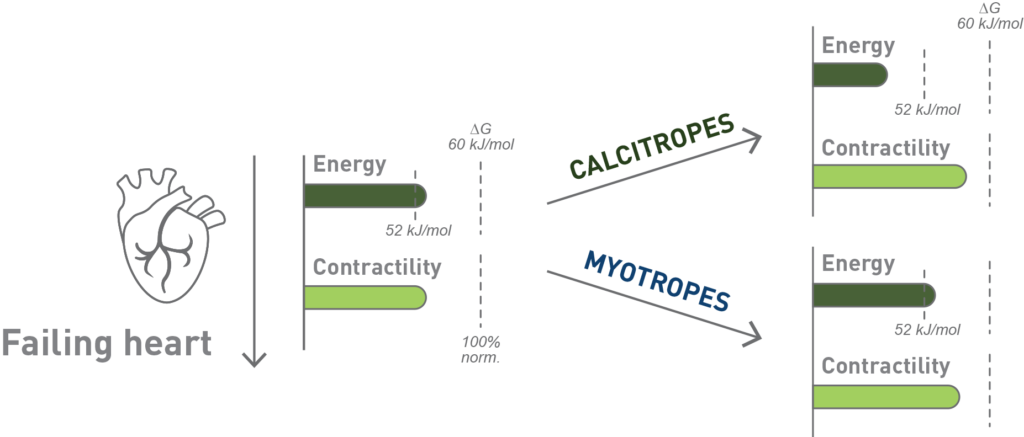
- Mitotropes target the energy dependence of myocardial contraction and the metabolic deficiencies present in the myocardium of patients with HFrEF2
- Myotropes target the sarcomere’s contractility via myosin, actin, and associated regulatory proteins through calcium-independent mechanisms2